TRANSLATION
第6回 「臨床試験の一般指針」に見る臨床開発の進め方
前回は治験を主に倫理的に適正に行うための手順を「臨床試験の実施の基準」(GCP)に則して述べました。今回は治験を科学的に適正に行うための方法論を定めた「臨床試験の一般指針」に沿って臨床開発の進め方を見ていきましょう。
臨床試験のガイドラインには、臨床試験全般の方法論を述べた「一般指針」のほか、臨床試験を様々な側面からみた指針があり、例えば「統計的原則」、「用量-反応関係」、「対照群の選択」などに関する指針、報告書のまとめ方に関する「治験の総括報告書」のガイドライン、さらに国際共同治験やブリッジングに関わる「外国臨床データの受け入れ」など多様です。また「抗不整脈薬」「鎮痛消炎薬」「抗高血圧薬」など個々の薬効群別に試験のデザインや注意事項などを述べたガイドラインもあります。
これらのガイドラインは日・米・欧3極間で協調して臨床開発を行い、データの相互受け入れを可能にするためにICHで合意されたものです。日本のガイドラインもこれに基づいていますので、ICHの英文も参照して下さい。(http://www.pmda.go.jp/ich/e/e8_98_4_21.pdf)
今回は主に「一般指針」に基づいて臨床試験の「相」という段階的な概念と各相で行われる試験の種類について見ていきましょう。関連する法律や用語については「治験ナビ」(治験メニュー→法律、ガイドライン、又は治験用語集など)も参照して下さい(時々誤字などがありますので、注意してください)。 (http://www.chikennavi.net/index.htm)
1. 臨床開発の進め方(4つの段階=相phase)
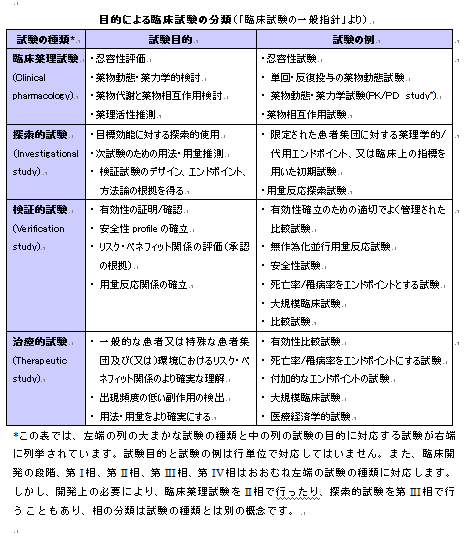
臨床開発は便宜的にI、II、III、IV相(phase I, phase II, phase III, phase IV)と4段階に分けて考えられています。治験は、各段階の目的に適うようにデザインされた治験実施計画書(protocol)に基づいて実施され、結果が解析され、それに基づいて治験総括報告書(clinical study report)が作成されます。つまり、いきなり大規模な比較試験を行うのではなく、前段階の結果を踏まえて試験の目的が設定され、課題が一つずつ、順にクリアされていきます。開発段階ごとに求められている試験を表に示します(文末の表入る)。試験の種類は開発される薬の性質や適応によって変わりますので、どの薬でもこの全てが実施される訳ではありませんが、その数は相当なものになります。
また、試験には「このように想定してこのような方法で行うとこのような結果が得られるはず」というパターン、すなわちデザインが必要です。試験目的にあわせて、デザインを選びます。治験実施計画書や治験総括報告書の標題部分などは、どのような試験であるかが、まるで「ジュゲムジュゲムゴコウノスリキレ…」といった調子で羅列されています。例えばA phase I open-label, single-dose treatment in healthy male Japanese subjects to determine bioavailability of XXXなど。治験の「相」とそこで行われる試験やデザインについて理解していると、このようなタイトルもどの相のどのような試験なのか、ある程度推測することができます。それでは、各相で行われる試験を見ていきましょう。
2.第I相(Phase I)で行われる試験(主に臨床薬理試験)
第I相は治験薬を初めて人に投与する段階で、通常、治療効果の確認はしません。まず、被験薬の忍容性(tolerability)と安全性を確認するために、少数の健常人や患者に被験薬を単回、次いで反復投与し、どこまで投与に耐えられるかを調べ、また予期される副作用の性質を判断して安全性を確認します。
次に薬物動態(pharmacokinetics)、薬力学(pharmacodynamics)、薬物相互作用(drug-drug interaction)などの基本的な問題点を確認して、これらの結果を次の段階の用量設定の根拠とします。薬物動態試験では、血中の薬物濃度や尿、糞などから被験薬(investigational drug)の体内動態(biological fate)(すなわち、吸収、分布、代謝、排泄)を推測し、クリアランスから未変化体又は代謝物の蓄積がないかを検討します(蓄積があると、薬効が強く出すぎたり、副作用が出たりします)。また、薬物は血球や目的外の組織と結合していると、目的とする組織で効果を発揮できませんので、投与量のうちどれくらいが血中にあって遊離(free)薬物として利用できるか生物学的利用率(バイオアベイラビリティ(bioavailability)も調べて、用量設定の参考とします。また、薬物は代謝されると、本来の作用を失うものが多いため、薬物代謝試験(drug metabolism study)で、血中・尿中等の代謝物の同定・測定から代謝経路や代謝の程度を調べます。中には体内で代謝されて初めて有効成分となる薬(プロドラッグprodrug)もあります。一方、薬物代謝酵素は民族によって異なる場合があり、民族差(ethnic difference)の有無を調べてその後の試験で海外データが利用できるかを検討します。これはまた、国際共同治験(global study)が可能かどうかの根拠ともなります
同じ薬物受容体に結合する複数の薬を同時に投与すると、受容体との結合部位で競合が起こり、その結果薬理効果が減少する場合があり、これを薬物相互作用といいます。そのような薬物の併用が予想される場合にはこの試験の実施が必要です。また、食事の影響が考えられる場合には食物との相互作用を検討する場合もあります。
薬によって、また治験のエンドポイント(治験行為の意義を評価する「評価項目」)によっては、被験薬による薬理反応の強度を生体内薬物濃度と関連付けて検討する薬物動態/薬力学試験(pharmacokinetic/pharmacodynamic study = PK/PD study)を健常人または目標疾患の患者を対象に行い、薬物濃度とともに臨床的指標(真の/代替エンドポイント、又は臨床効果や副作用との関係が確認された臨床薬理学的指標)を評価します。
通常第I相で行われる薬物動態や薬力学の試験をひっくるめて臨床薬理試験(clinical pharmacology study)といいます。しかし、既に述べたように「相」という概念は便宜的なもので、ここに述べた試験でも場合によっては第II相で行われることもあります。例えば第II相で新たな併用薬が候補に上がれば、その時点で薬物相互作用試験の必要性が生じます。
3.第II相(Phase II)で行われる試験(主に探索的試験)
第II相では患者における治療効果を探索する探索的試験(exploratory study)を開始します。探索的試験では、まず同時対照群(並行比較群ともいう、parallel control)や投与前の状態(baseline)との比較を行い、次に特定の適応(indication)に対する有効性(efficacy)と安全性(safety)の検討を比較的均質な少数の患者を対象に、無作為化同時対照比較(randomized parallel control)により行います。
ところで、対照(control)とは何でしょうか。これは言葉で、被験薬(新薬候補として試験する薬、investigational drug)と比較する対象をいいます。対照薬(control, control drug)にはプラセボ(外観は被験薬と同じで有効成分の入っていない薬。通常、乳糖などplacebo)と実薬(有効成分の入っている薬、多くは市販薬、active drug)があります。被験薬と対照薬を合わせて治験薬(study drug)といいます。対照薬使用の有無、対照薬の種類、投与方法、評価方法などによって試験のデザインが変わってきます。
また、無作為化(randomization)とは、いくつかある治療法のひとつに被験者を無作為に(ランダムに)割り当てることによって、恣意的に特定の治療群に割り当てる(例えば治療効果の高そうな被験者を被験薬群に割り当てる)バイアスを避けることをいいます。
第II相の主な目的は第III相試験の用法・用量を決定することで、初期には用量漸増デザイン(dose-escalating design)、次いで並行用量反応デザイン(parallel group dose-response design)が用いられます。それぞれの試験で使われる主なデザインはその試験のガイドライン(例えば「用量-反応関係の検討のための指針」)に詳しく解説されています。デザインについては「治験ナビ」の用語集にも簡単に解説されています。
また、第II相では、その後に実施される第II相や第III相試験において用いられる見込みのあるエンドポイント・治療法(併用療法を含む)・対象患者群 (重症度severityによる分類など)などの評価も行われます。用語は、治験実施計画書や治験総括報告書の目次の次にglossaryがついていることが多いので、それを参照することもできます。
4.第III相(Phase III)で行われる試験(主に検証的試験)
第III相では、通常、治療上の利益の証明または確認を目的とする検証的試験(verification study)試験を行います。つまり第II相で蓄積された予備的な証拠(preliminary evidence)「被験薬は意図した適応及び対象患者群において安全で有効である」ということを検証します。ここではかなり大規模な患者数(場合によっては数千人)で、市販薬を対照として、想定される臨床使用状況を考慮して試験が行われます。ここで得られたデータがこの薬の承認の根拠となります。
また、場合によっては、用量-反応関係のさらなる探索、より広い対象患者や病態の異なるステージでの被験薬の使用、他剤との併用(combination therapy)などが検討され、長期投与の予想される薬では、長期試験(long-term study、場合によっては延長試験extension study)、高齢者への投与が想定されている場合は高齢者を対象とした試験も行われます。また、妊婦・授乳婦、小児への投与が予想される薬物では、これらの集団(population)を被験者とした試験を考慮します。
第III相試験で得られるこれらの情報はその薬の正式な製品情報として、後に添付文書(package insert)に記載されます。
5.第IV相(Phase IV)で行われる(治療的使用)
第IV相の試験は、その薬の承認後に(つまり市場に出てからpost-marketing)行われる試験です。開発段階で安全性、有効性が確認され、用量が設定されてはいるが、実際の治療環境でそれ以上の知見を得るために行います。
これは定期報告が求められている承認販売後調査を除く全ての承認販売後臨床試験が含まれ、追加的な薬物相互作用試験、用量-反応試験、安全性試験、承認された適応疾患における使用を支持する試験などが含まれます。
新効能、新用法・用量、新投与経路、追加の患者集団での試験は新たな開発計画のもと、別途行われます。
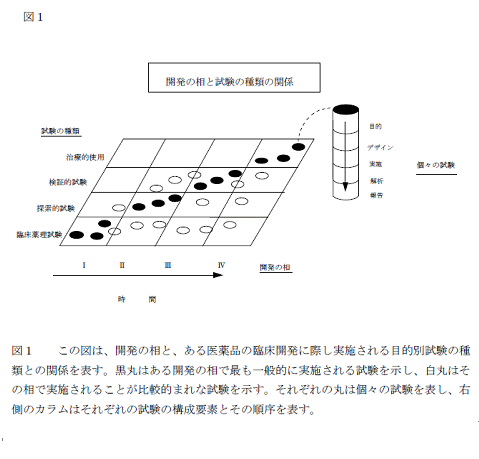
治験の各相でどのように試験が行われるかが下図に示されています。(図入る)これを見ると、開発が進むにつれ、その時点での必要性、目的に応じて試験が選択されることがわかります。どの試験は必ずどの相でと決められているものではないのです。初めに「相」は便宜的な概念だと書いたのは、このように「相」があって開発が行われるのではなく、それぞれの薬に応じて必要な試験が選択されるからです。相はむしろ概念なのです
次回はこのようにして行われた臨床試験の結果をどのような形にまとめるのか、「治験の総括報告書」を中心にお話します。