TRANSLATION
第5回 臨床試験(1) GCPと治験の実施手順
今回は人を対象とした試験、すなわち臨床試験に入ります。サイエンスとしての「臨床試験」という用語と薬事規制上の用語である「治験」については第1回で説明しました。以下で「臨床試験」という時、もちろん「治験」もここに含まれるとお考えください。
治験の目的は人を対象として薬の有効性と安全性を確認することですが、その実施にあたって、最も留意すべきことは何でしょうか?それはまず、被験者の人権が保護され、倫理的で安全、なおかつ、科学的に妥当な方法で試験が実施されることです。倫理性と科学的妥当性は車の両輪のようなもので、科学的に斬新であっても、倫理的に妥当な方法で試験が行われなければ、被験者の安全や人権は守られず、人を対象とした科学の方法としては適切とはいえません。一方、真に科学的な方法で試験が行われなければ、その結果は偽りとなり、被験者の協力も無駄となります。
治験に対する法規制には、主にその手続き面を定めた基準と、科学的方法を定めたガイドラインとがあります。今回は「臨床試験の実施の基準」(GCP)を通して、治験の手順を見て行きましょう。
1. 医薬品の臨床試験の実施の基準
新薬の製造販売承認申請には、その品質、有効性、安全性を確保するための多方面の資料が求められ、そのための試験が行われます。臨床試験もそのひとつですが、その適正な実施のための「手続き面」を定めたものが「医薬品の臨床試験の実施の基準」(Good Clinical Practice = GCP)です。詳細は下記を参照して下さい。ICHのGCP(英文)も参照できます。
GCP: http://law.e-gov.go.jp/htmldata/H09/H09F03601000028.html
*基準としてはGCP以外に、分野によって「医薬品の安全性に関する非臨床試験の実施の基準」(Good Laboratory Practice = GLP)、「医薬品の製造管理及び品質管理に関する基準(Good Manufacturing Practice = GMP)、「製造販売後の調査及び試験実施基準」(Good Postmarketing Study Practice = GPSP)などがあります。
臨床試験は過去にナチスや日本軍による人体実験や、自由意思を表明できない囚人等を被験者にするなど、倫理面での負の遺産が多数ありました。これらの反省に立って、2度と非倫理的な臨床試験を行わないことを医師達が誓ったものがヘルシンキ宣言(Declaration of Helsinki) ( http://www.mext.go.jp/a_menu/shinkou/sangaku/gijiroku/04090601/010.pdf)です。GCPはその精神に則り、患者の自由意思に基づいた倫理的で科学的な臨床試験を行うための手続きを定めたものです。倫理的・科学的妥当性は当然個々の試験の治験実施計画書に盛り込まれ、治験依頼者や医師が精神としてそのような治験実施を心がけることは当然ですが、GCPはここに定めた手続きを踏むことによって、システムとして適正な治験の実施を目指すものです。GCPと治験に関しては、上記サイトのほか、関連の法令、用語集なども含めて参照できるものとして治験関連サイト「治験ナビ」があり、翻訳者にとって参考になります。(http://www.chikennavi.net/i_gcp_index.htm)
2. GCPと治験の手順
治験はどのような人や組織によって、どのような手順で実施されるのか、GCPの記述に基づき、治験の流れをごく大まかな図にしました。○の数字を追っていくと、主な手順がわかります。ここには主にピンク色で「治験実施」業務の流れ、緑色で「監査」業務の流れ、青色で「有害事象発現の場合」の業務の流れが示されています。治験はこのように様々な人や組織が関与する壮大で総合的なプロジェクトなのです。
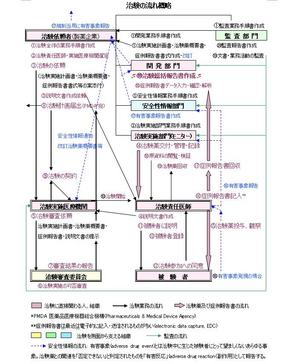
▶図をクリックして拡大
上記手順の主な登場人物・組織とその業務、及びそこで作成される文書等を以下に記述します。用語はICHのGCPの英語に日本の治験で従来使われていた日本語を対応させているため、考え方や医療環境の違いから、英訳・和訳ともに直訳では思い浮かばない表現が多く、要注意!です。詳細はICHのGCPと日本のGCPを対比して見てください。ここでは企業主導の治験について説明します。(ほかに「医師主導の治験」があり、「治験依頼者」の代わりに医師が自ら治験を実施する点で、責務、手順等が異なります。)
1)治験依頼者(Sponsor)
多くは製薬企業で、業務手順書(standard operation procedure=SOP)に基づき、治験実施医療機関及び治験責任医師を選定し、治験実施契約を結ぶ。製薬企業には通常、臨床開発 (Clinical Development) 部門、治験実施 (Clinical Research Operation) 部門、安全性情報 (Drug Information) 部門、監査部門等があり、以下の業務を担当する。
臨床開発部門:開発計画の策定、治験実施計画書(protocol)・治験薬概要書(investigator’s brochure)の作成・改訂、症例報告書(case card)書式の作成。症例報告書データの確認・入力・統計解析(statistic analysis)とこれに基づく治験総括報告書(clinical study report)の作成。
治験実施部門:治験薬(study drugs=被験薬investigational drug + 対照薬control drug)の管理(出庫・回収・保管・記録)とモニター(monitor)による交付・回収、治験実施中の被験者の人権・安全等の保護及び治験実施計画書への準拠・GCP遵守等についてのモニタリング、症例報告書 (case report form) データとカルテ等の原資料(source data)との照合・検証(validate)。
安全性情報部門:通常の治験実施には関わらないが、有害事象(adverse drug event)
発現の場合、治験薬との因果関係(関連が否定できなければ、有害反応adverse drug reaction=副作用)などを検討し、有害事象報告書を作成。
監査部門(Audit):会社自体の監査部門と区別して、信頼性保証(Quality Assurance)部門とも呼ばれ、上記の諸部門とは独立の立場で、治験に関わる全ての業務活動及び文書をGCPその他の規制要件に照らして監査(audit)する。
なお、実施部門又は開発部門の業務の一部を外部の治験業務受託機関(contract research organization=CRO)に委託することがある。
2)治験実施医療機関(Institution, 個々の施設についてはtrial site, stationともいう)
十分な臨床観察・試験検査を行う設備と人員を有し、被験者に対する緊急措置が可能で、治験審査委員会(investigational review board, IRB)が設置され、十分数の治験責任医師および治験協力者(薬剤師、看護師、治験コーディネーターclinical research coordinator=CRC)等を有する医療施設。
なお、日本では治験依頼者と治験実施医療機関が契約を結ぶが、海外では治験依頼者と治験責任医師が直接契約を結ぶので、手順が異なる。
3)治験責任医師(Investigator)
治験実施にふさわしい教育・訓練を受け、十分な臨床経験があり、治験実施計画書・治験薬概要書に定める治験薬の使用法に精通している医師を治験依頼者が選定。治験分担医師、治験協力者と業務分担する場合の責任者。治験について被験者に説明し、参加同意を得る。治験実施計画書に従って治験を行い、症例報告書を作成。有害事象を治験依頼者に報告。
4)治験審査委員会(Institutional Review Board)
治験実施の適否を評価するため、実施医療機関ごとに設置される委員会。治験について倫理的・科学的見地から審議する。第3者委員を含み、医療・臨床の専門家以外の者を含むこと。
次回は臨床試験のガイドラインに沿って、治験のデザインや相(フェーズ)について解説いたします。

