TRANSLATION
第9回 治験薬概要書及びCTDに関する概説
本講座の第7回、第8回で取り上げた「治験の総括報告書」は一つの治験の総まとめで、いわば苦労を重ねて実施してきた治験の花道とも言えます。折角ここまでたどり着いたのに、話は少し戻りますが、この治験の過程で作成される重要な文書として「治験薬概要書(investigator’s brochure, IB」があります。今回はこの「治験薬概要書」と、後に全ての治験終了後に作られる「コモンテクニカルドキュメント(Common Technical Document, CTD)」について、概説します。
この2つは、作成時点も目的も全く異なりますが、翻訳者から見た文書の性質としては「それぞれの作成時点までの非臨床から臨床までの全ての試験を網羅した概説」という点が似ています。それまでの試験のReviewであるため、用語や試験内容という点では、初めて毒性試験なり臨床試験なりに向き合った時のように、目新しい用語や試験方法が出てくるわけではありません。ですので、それまでに非臨床・臨床試験の翻訳で蓄積してきた知識を踏まえて訳すことになります。
とはいえ、これらの文書の目的や、その構成を知っていれば、訳す際に参考になりますので、今回はその構成を中心に、ごく簡単に説明します。
1.治験薬概要書
治験薬概要書は治験責任医師(investigator)や治験協力者に向けた、治験薬についての臨床及び非臨床データ(clinical and non-clinical data)のまとめで、被験者の臨床的管理に必要な情報の提供を意図しています。「医薬品の臨床試験の実施の基準(GCP)」は治験依頼者(sponsor)が「規定の試験により得られた資料並びに被験薬の品質、有効性及び安全性に関する情報に基づいて、以下の事項を記載した治験薬概要書を作成すること」を求めています。
1) 被験薬の化学名(chemical name)又は識別記号(development code)
2) 品質(quality)、毒性(toxicity)、薬理作用(pharmacology)その他の被験薬に関する事項
3) 臨床試験が実施されている場合は、その試験成績
本講座第5回の「治験の流れ」の図に示したように、治験依頼者は治験実施依頼の際に、治験実施計画書とともに治験薬概要書を作成して、治験実施医療機関に提出します。治験実施医療機関では、治験審査委員会(Institutional Review Board, IRB)が治験薬概要書と治験実施計画書(protocol)に基づいて当該治験実施の可否を審査し、その承認に基づき、治験が実施されます。治験責任医師、治験分担医師は治験薬概要書の内容を完全に理解し、熟知した上で治験を行い、また、 治験ナース、治験コーディネーター(clinical research coordinator、CRC)などの治験協力者も必要に応じて治験薬概要書を参照します。さらに、治験依頼者が当局に治験届書を提出する際にも、治験実施計画書と治験薬概要書を添付します。
治験依頼者は被験薬の品質、有効性及び安全性に関する事項のほか、治験を適正に行うために重要な情報を入手した時は、治験薬概要書を改訂します。例えば国内又は海外の治験で新たな副作用が報告された、あるいは海外の治験で新たな知見が得られた、海外でア新たな規制が適用された、などの場合です。
以下はあるパーキンソン病治療薬の治験薬概要書の目次の例です。このように、Introduction(当該薬剤の開発の目的や経緯)から始まり、非臨床、臨床とそれまでに得られた結果を概説していきます。どのような内容が含まれるのか、一例としてこれを訳してみましょう。
SUMMARY
1. INTRODUCTION
2. PHYSICAL, CHEMICAL AND PHARMACEUTICAL PROPERTIES
2.1. NOMENCLATURE
2.2. DESCRIPTION
2.3. PHARMACEUTICAL DATA
3. PRECLINICAL STUDIES
3.1. PRECLINICAL PHARMACOLOGY
3.2. PHARMACOKINETICS IN ANIMALS
3.3. TOXICOLOGY
4. EFFECTS IN HUMANS
4.1. PHARMACOKINETICS OF XXXX
4.1.1. Studies in healthy volunteers
4.1.2. Studies in special populations
4.1.3. Studies in patients with Parkinson’s disease
4.2. PHARMACODYNAMICS AND CLINICAL EFFICACY OF XXXX
4.2.1. Phase I studies
4.2.2. Phase II studies
4.2.3. Phase III studies
4.3. TOLERABILITY AND SAFETY OF XXXX
4.3.1. Phase I studies
4.3.1.1. Adverse events and laboratory parameters
4.3.1.2. Serious adverse events
4.3.2. Phase II studies
4.3.2.1. Adverse events and laboratory parameters
4.3.2.2. Serious averse events
4.3.3. Phase III studies
4.3.3.1. Adverse events
4.3.3.2. Serious adverse events
4.3.3.3. Premature treatment withdrawals
4.3.3.4. Laboratory parameters
4.3.3.5. Tolerability and in-patient subgroups
4.3.3.6. Effects of concomitant medication on tolerability
4.3.3.7. Overdose
4.3.3.8. Withdrawal of XXXX
4.3.4 Post-marketing experience
5. GUIDANCE FOR THE INVESTIGATOR
6. REFERENCES
(訳)
要 約
1. 緒言
2. 物理・化学及び製剤学的特性
2.1. 名称
2.2. 性状
3.3. 製剤学的データ
3. 前臨床試験
3.1 前臨床薬理試験
3.2 動物における薬物動態
3.3. 毒性試験
4. ヒトにおける効果
4.1. XXXXの薬物動態
4.1.1. 健常人を対象とした試験
4.1.2. 特別な母集団を対象とした試験
4.1.3. パーキンソン病患者を対象とした試験
4.2. XXXXの薬力学及び臨床効果
4.2.1. 第I相試験
4.2.2. 第II相試験
4.2.3. 第III相試験
4.3. XXXXの忍容性と安全性
4.3.1. 第I相試験
4.3.1.1. 有害事象と臨床検査値
4.3.1.2. 重篤な有害事象
4.3.2. 第II相試験
4.3.2.1. 有害事象と臨床検査値
4.3.2.2. 重篤な有害事象
4.3.3. 第III相試験
4.3.3.1. 有害事象
4.3.3.2. 重篤な有害事象
4.3.3.3. 治療からの早期脱落
4.3.3.4. 臨床検査値
4.3.3.5. 入院患者サブグループの忍容性
4.3.3.6. 忍容性に対する併用薬の影響
4.3.3.7. 過量投与
4.3.3.8. XXXXの投与中止例
4.3.4. 販売承認後の経験
5. 治験責任医師のための指針
6. 文献
治験薬概要書の内容は開発品目や治療領域、開発段階などによって、当然変わってきます。また各項目は必要に応じてさらに細分化されます。治験薬概要書は開発計画の問題点の検討も兼ねて製薬会社内で訳す場合も多いのですが、分量が多いため、外注されることも結構あります。この文書は非臨床から臨床まで広範囲にわたってその品目について知ることができ、じっくり取り組む時間があれば、大変面白いし、勉強になります。しかし、納期が短いことも多いので、そのような場合は、手分けしてそれぞれの分野を得意とする翻訳者が訳します。この場合は訳者間の用語の統一方法を決めておきます。注意しなければならないのは、これは「概要書」なので、各試験の方法やデータなどがかなり割愛されていて、事実や論理展開が把握しにくい場合があることです。そのような場合は発注元の会社に問い合わせるなり、関連部分のデータを見せてもらいましょう。それができない場合には、納品時に疑問箇所とその理由を明記しておきます。
2. コモンテクニカルドキュメント(CTD)
前回、前々回の「治験の総括報告書」は一つの治験についての総まとめでした。このような臨床試験が第I相から第III相まで数多く積み重ねられ、さらにそれ以前には非臨床の試験も多数行われてきて、いよいよ、これら全てをまとめて、新薬の承認申請を行います。
開発過程で得られたデータはコモンテクニカルドキュメント(CTD)の様式でまとめられ、申請書とともに規制当局に提出されます。CTDとは、ICHでの日・米・欧の3極合意に基づいた共通の様式で、この様式でまとめることより、日米欧いずれの地域の当局にも受け入れられます。なお、CTDは国際的に用いるこが前提なので、英文にしておく必要があります。Global企業では、英文版を正本とし、日本語版を副本とする会社が多いかと思いますが、これをFDAなどに提出する場合には、「英文版と日本語版の内容が全く同じである」という証明(陳述)が求められます。
CTDのガイドラインは医薬品医療機器総合機構のICHのサイトを参照して下さい。
(http://www.pmda.go.jp/ich/ich_index.html
ここから「複合領域」をクリックすると、CTD関連の資料が色々検索できます。
CTDの構成は以下のような階層構造で表されます。これは上から1, 2 ,3の番号がついていますが、開発過程としては底辺の第3部、第4部、第5部の試験結果に基づいて第2部の品質、非臨床、臨床の概括評価が行われ、最終的に第1部の申請資料目次がまとめられ、添付文書情報などに反映されて、申請に至るというプロセスを辿ります。
また、各項目の項立て、項目番号も3極で共通化されています。
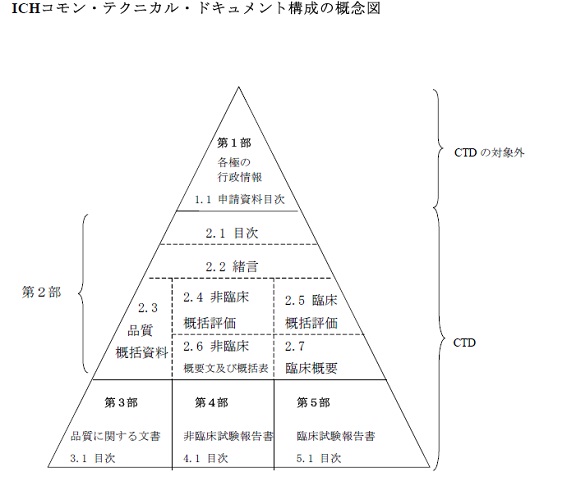
では、各モジュールの内容を見ていきましょう。
第1部(Module 1) 申請書等行政情報及び添付文書に関する情報
第1部には各極特有の申請書及び行政情報(regional administrative information)や添付文書情報(package insert information)が記載されます。翻訳の仕事としては、行政情報や当局とのやりとりはCTDのまとめの際よりも、Global開発の調整のため、開発途中でその都度行われ、どちらかといえば英訳が主です。
添付文書(案)はその医薬品の成分、効能・効果、用法・用量、使用上の注意、など、いわばその薬の最終的なプロフィールを表しているもので、承認された暁には、医師や患者との接点になる文書です。製薬会社はこれに対する承認を得るために、努力を重ねているともいえます。承認後も副作用情報や使用上の注意などの改訂が行われますので、和訳も英訳も翻訳需要はあります。添付文書については次回詳しく述べます。
第2部(Module 2) CTDの概要(Common Technical Document Summaries)
CTDの翻訳の仕事としてはこの部分が多いかと思います。この部分の構成は以下の英文の通りです。これを和訳してみましょう。
(例題)

(訳)
第2部(モジュール2) CTDの概要(サマリー)
第2部(モジュール2)は、薬理学的分類、作用機序及び申請する効能又は効果等の当該医薬品の全般的な概略から始めること。原則として、この緒言は1ページ以内にまとめること。
第2部(モジュール2)は、以下の順番で7項目を含むこと:
• 目次
• 緒言
• 品質に関する概括資料
• 非臨床に関する概括評価
• 臨床に関する概括評価
• 非臨床試験に関する概要文及び概要表
• 臨床概要
この訳は実は日本語のCTDガイドラインの表現そのままで、直訳ではありません。OverviewやSummaryなど、似たような英語が出てきて、その日本語がまた概括資料、概括評価、概要と似たような言葉が並び、これは言葉だけ見ていても分かりにくいですね。ガイドライン本文を見ると、下記のように、指しているものが少しずつ違うのです。
「品質に関する概括資料」(Quality overall summary, QOS):第3部の資料をその範囲及び構成に即して要約したもの。
「非臨床に関する概括評価」(Non-clinical overview):当該医薬品の薬理、薬物動態及び毒性についての包括的で客観的な評価結果。最初に「非臨床試験計画の概略」を示し、次いで「薬理試験」、「薬物動態試験」、「毒性試験」の順に、行った試験とその結果及び意義を述べ、最後に「総括及び結論」で当該医薬品の特徴を明確にし、目的とする臨床使用における安全性の裏づけを述べます。
「非臨床試験に関する概要及び概要表」(Non-clinical written and tabulated summary):行った非臨床試験について、ガイドラインの記載要領に沿ってさらに詳述(100~150ページ)するもので、全体を比較できるような概要表(規定の形式)も入ります。
「臨床に関する概括評価」(Clinical overview):当該医薬品の臨床的意義の審査に用いられ、「臨床概要」、個々の「治験総括報告書」その他関連資料から得られた結論を評価します。開発計画及び試験結果の優れた点と限界を示し、また、試験結果が添付文書中の記載にどのように反映されているか、記述します(約30ページ)。内容の詳細は、製品開発の根拠(rationale for the product development)、生物薬剤学(biopharmaceutics)・臨床薬理(clinical pharmacology)・有効性(efficacy)・安全性(safety)等に関する概括評価、既存の治療法との比較、ベネフィットとリスクに関する結論(risk/benefit analysis)などを含みます。
「臨床概要」(Clinical summary):CTD中の全ての臨床情報の詳細な事実に基づく要約の提供。第5部の「治験の総括報告書」や、メタアナリシスや複数試験の併合解析、他の地域の市販後データなどの情報も含んだ、事実に基づく観察、結果の記述(50~400ページ)。やはり生物薬剤学、臨床薬理、臨床的有効性、臨床的安全性の各分野のデータを取り上げ、分野ごとの全試験を通しての結果の比較と解析を記述します。臨床的安全性の分野では、特別な患者集団や状況下での分析、海外の市販後データなども取り上げます。
第3部(module 3): 品質に関する文書(Documents on quality)
品質に関する資料で、原薬、製剤、規格、製造方法等に関する報告書/データ等の資料をガイドラインの記載要領に従って記載したもの。
第4部(module 4):非臨床試験報告書(Non-clinical study reports)
実施した非臨床試験を薬理試験、薬物動態試験、毒性試験の順に配列し、ガイドラインの規定に沿って図表なども取り入れて記述したもの。
第5部(module 5):臨床試験報告書(Clinical study reports)
実施した全ての治験の総括報告書を一覧表で示し、これらをガイドラインの配列順、すなわち、生物薬剤学、ヒト生体資料を用いた薬物動態、臨床薬物動態(PK)、臨床薬力学(PD)、有効性及び安全性、の順に配列し、また、(他地域などの)市販後のデータ及び患者データ一覧表及び症例記録を添付したもの。
これでお分かりのように、CTDは実に膨大な資料になり、print-outを積み上げると数メートルになります。このような膨大なハードコピーを扱うのは大変で、データの記録、伝送、まとめの段階から電子化しておけば、検索、参照、保存、検証、様々な面で便利で確実です。そのため、現在では、治験中からelectronic data capture (EDC)システムを導入してデータを記録し、さらに治験総括報告書もeCSRとしてまとめ、最終的にCTDもeCTDの形で提出する会社が多くなっています。

